|
Few representative studies
(1) Ab initio density functional theory based methods are employed to study the reactivity of various kinds of metallic, bimetallic, and metal carbide clusters towards the adsorption activation of CO2 molecule to assess the catalytic efficacy of these clusters in the dissociation of CO2.
(a) Adsorption and Activation of Carbon dioxide molecule on small Zirconium clusters
The elevated levels of carbon dioxide (CO2) is a matter of great concern as it has been considered as one of the gases which might contribute to the global warming through the greenhouse effect. The recycling of CO2 into other useful chemicals is one of the practical ways to tackle this problem as CO2 is very abundant and it will also help to fight against the fuel crises. The first step for such process is to activate CO2 using catalysts. To this end, adsorption and activation of CO2 on small sized Zrn (n=2-7) clusters using density functional theory have been studied. From our calculations we find that there is very high adsorption and activation of CO2 molecule on all the Zrn clusters which is reflected in the elongation of C-O bond length (C-O>1.27 Å) and the deviation of bond angle O-C-O (vary between 114º and 136º) from the respective values of gas phase CO2 molecule (C-O bond length is 1.160 Å and the bond angle O-C-O is 180.0°). The high activation of C-O bond is also attributed to the significant charge transfer from cluster to the CO2 molecule. The calculations for the transition state (TS) search predict that the energy barrier for dissociation of CO2 to CO and O is very small for all the clusters except Zr2 and Zr4 clusters. From this study it is clear that Zr clusters may serve as highly efficient catalyst for the hydrogenation of CO2 to useful fuels.
![Figure 1: (a) Optimized structures of reactant ([A]), TS, and dissociated product ([B]) and (b) Reaction path for CO<sub >2</sub> dissociation to CO and O on Zr7 cluster.](img/tsl_pic_1.PNG) Figure 1: (a) Optimized structures of reactant ([A]), TS, and dissociated product ([B]) and (b) Reaction path for CO2 dissociation to CO and O on Zr7 cluster.
Figure 1: (a) Optimized structures of reactant ([A]), TS, and dissociated product ([B]) and (b) Reaction path for CO2 dissociation to CO and O on Zr7 cluster.
(b) Adsorption and Activation of Carbon dioxide molecule on Small Sized Bimetallic Cu-Zr clusters
Adsorption and activation of CO2 molecule on small sized bimetallic Cu 4-nZrn (n = 1-4) clusters using density functional theory have been studied. From our calculations we find that there is very high adsorption and activation of CO2 molecule on all the Cu-Zr teranuclear clusters except for monometallic Cu4 which is also reflected in the elongation of C-O bond length (C-O >1.26 Å) and the deviation of bond angle O-C-O (vary between 116º and 137º) from the respective values of free CO2 (C-O bond length is 1.160 Å and the bond angle O-C-O is 180.0°). Furthermore, we find that the adsorption energy of CO2 can be tuned by varying the number of Zr atoms in the Cu−Zr clusters. The high activation of C-O bond is also attributed to the significant charge transfer from the cluster to the CO2 molecule. The calculations for the transition state (TS) search predict that the energy barrier for dissociation of CO2 to CO and O can be reduced by incorporating the Zr atoms in the Cu4 cluster. In particular, the energy barriers are considerably small for Cu3Zr and CuZr3 clusters. From this study it is clear that Cu3Zr and CuZr3 clusters may serve as highly efficient catalyst for activation and dissociation of the CO2 molecule.
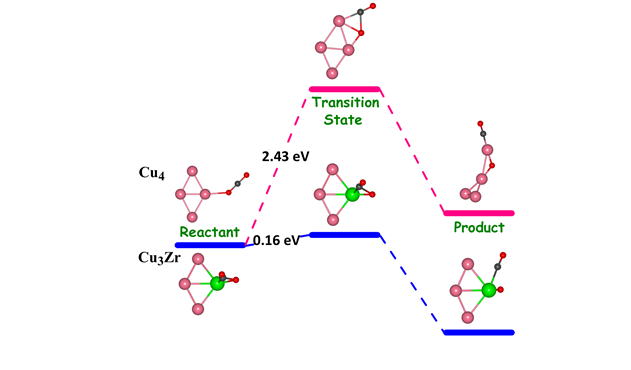 Figure 2: Reaction pathways showing the significant reduction in CO2 dissociation barrier with incorporation of Zr atom.
Figure 2: Reaction pathways showing the significant reduction in CO2 dissociation barrier with incorporation of Zr atom.
(2) Adsorption and Activation of CO2 molecule by Metcars
(Metallocarbohedrenes, more commonly known “metcars” are very stable metal carbide clusters with chemical stoichiometry M8C12, where “M” is usually an early “d” block element. The efficacy of metcars like Ti8C12 and Nb8C12 towards adsorption, activation, and dissociation of CO2 molecule has been explored by employing ab initio density functional theory based method. To determine the structures, detailed geometry optimization calculations have been carried out using three different exchange-correlation functionals of ascending order of accuracy and three basis sets of increasing size. Among several isomers, we find that the isomer of Ti8C12 with C3v point group symmetry is the most stable one and the isomer with Cs symmetry is found to be the lowest energy structure for Nb8C12 metcar. Further, our results suggest that the CO2 molecule is strongly adsorbed and undergoes a significantly high degree of activation onto both the Ti8C12 and Nb8C12 metcars. The migration of charge from Ti/Nb metcar to CO2 molecule is responsible for high degree of activation of this molecule. The results for the energy barrier for the dissociation of CO2 molecule to CO and O fragments on both of the Ti8C12 and Nb8C12 clusters revealed that the minimum barrier to be overcome is of the order of 1.0 eV
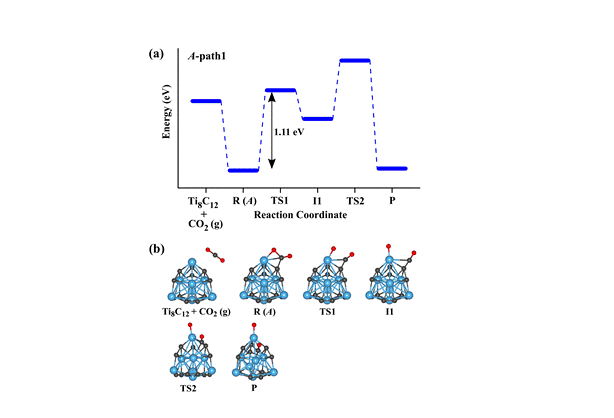 Figure 3. Reaction path for CO2 dissociation into CO and O fragments on a Ti8C12 metcar obtained using the B3LYP XC functional and LanL2DZ basis set.
Figure 3. Reaction path for CO2 dissociation into CO and O fragments on a Ti8C12 metcar obtained using the B3LYP XC functional and LanL2DZ basis set.
(3) Studies on 2D Dipolar spin-polarized Fermi gas
An analytically soluble model of two dipolar fermions trapped in a harmonic potential has been employed to assess the accuracy of the correlation energy functional derived within local density approximation. This expression for the correlation energy is used to study the effect of correlation on the ground-state and collective oscillations of a spin-polarized dipolar Fermi gas trapped in a two-dimensional harmonic potential.
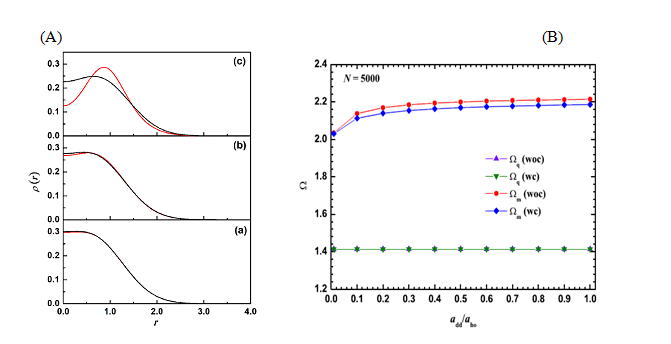 Figure 4(A) Comparison of exact density (black) with the ones obtained with Kohn-Sham calculations (red) for different values of dipolar interaction strength (a) add /l0 = 0.1, (b) add /l0 = 0.3, and (c) add /l0 = 1.0. (B) Variation of monopole (Ωm) and quadrupole (Ωq) with dipolar interaction add /l0 for N = 5000 fermions. “woc” and “wc” denote results without correlation and with correlation effects taken into account, respectively.
Figure 4(A) Comparison of exact density (black) with the ones obtained with Kohn-Sham calculations (red) for different values of dipolar interaction strength (a) add /l0 = 0.1, (b) add /l0 = 0.3, and (c) add /l0 = 1.0. (B) Variation of monopole (Ωm) and quadrupole (Ωq) with dipolar interaction add /l0 for N = 5000 fermions. “woc” and “wc” denote results without correlation and with correlation effects taken into account, respectively.
(4) Surface Termination and Thickness Dependent Magnetic Coupling of Cr Adlayers on
Ni2MnGa(001) Surfaces
In this work, we explore the magnetic properties of Cr adlayers on the (001) surface of a ferromagnetic Heusler alloy, namely Ni2MnGa, using density functional theory based electronic structure calculations. The magnitudes and orientation of the magnetic moments of the Cr adlayers on both the Mn-Ga and Ni terminated Ni2MnGa (001) surfaces have been investigated and we find that the magnetic ordering is sensitive to the substrate atoms and the adlayer thickness as well. For one monolayer (ML) of Cr adlayer on Mn-Ga (Ni) surface, the Cr atoms are ferromagnetically (anti-ferromagnetically) coupled within the layer which has been observed from the results obtained from crystal orbital Hamilton population (COHP) analysis (Figure 1) as well as Heisenberg exchange coupling constants between the surface and sub-surface atoms. However, for two monolayers of Cr, the Cr adatoms are ferromagnetically coupled, irrespective of the surface termination. Our results suggest weaker coupling of Cr adatoms with the Ni terminated surface as compared to the Mn-Ga case. Further, for the surface Cr atoms, significant enhancement of magnetic moments has been observed in all the cases. Our work is expected to give useful insight for future work on probing composite systems of thin adlayers on the surface of Heusler alloys, for understanding different aspects of two-dimensional magnetism.
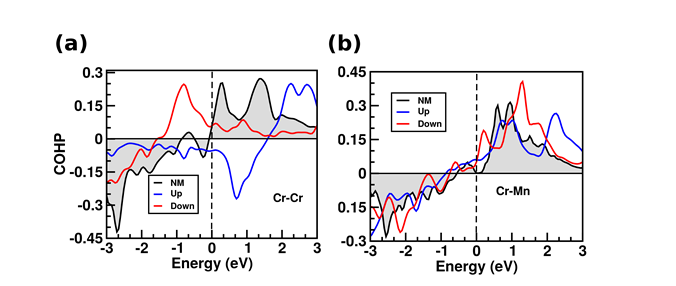 Figure 5. Validation of (a) anti-ferromagnetic coupling between surface Cr atoms and (b) ferromagnetic coupling between top Cr and sub-surface Mn atoms, respectively, in case of 1ML of Cr adlayer on Mn-Ga terminated surface.
Figure 5. Validation of (a) anti-ferromagnetic coupling between surface Cr atoms and (b) ferromagnetic coupling between top Cr and sub-surface Mn atoms, respectively, in case of 1ML of Cr adlayer on Mn-Ga terminated surface.
(5) Probing the Martensite transition and thermoelectric properties of CoxTaZ Heusler alloys (Z = Si, Ge, Sn with x = 1, 2)
Spin polarized electronic structure calculations have been carried out using density functional theory based method to systematically investigate the possibility of existence of the tetragonal phase in the full (FHA) and half (HHA) Heusler alloys, CoXTaZ (Z = Si, Ge, Sn with x = 1,2). A tetragonal phase has been found to be stable up to about 400K in case of Co2TaSi and Co2TaGe alloys and up to about 115K for Co2TaSn. The energetics and electronic structure results corroborate well with the elastic properties of these materials in establishing that the symmetry of the lowest energy state for the FHAs is indeed tetragonal, unlike the HHAs (Fig. 6). Further, it is found that the FHAs are magnetic and of metallic nature in both the phases, implying that these alloys belong to the group of magnetic shape memory alloys. Further, due to the semi-conducting nature of the respective HHAs, enhanced thermoelectric properties (by about an order) have been seen in this class of alloys (Fig. 7), which has been explained by the results of respective Fermi surfaces.
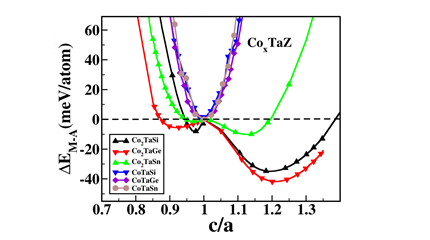 Figure 6 Energy difference between the austenite and martensite phase as a function of c/a for the CoxTaZ Heusler alloys
Figure 6 Energy difference between the austenite and martensite phase as a function of c/a for the CoxTaZ Heusler alloys
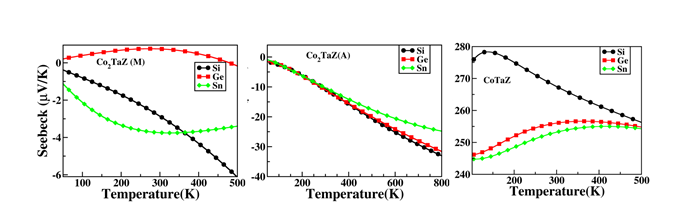 Figure 7 Seebeeck coefficients of CoXTaZ (X=1,2: Z =Si, Ge, Sn) alloys. A and M stands for the austenite and martensite phase, respectively.
Figure 7 Seebeeck coefficients of CoXTaZ (X=1,2: Z =Si, Ge, Sn) alloys. A and M stands for the austenite and martensite phase, respectively.
(6) Massless Dirac fermions in stable two-dimensional carbon-arsenic monolayer
A monolayer made up of carbon and arsenic atoms with a chemical composition (CAs3) is predicted to be energetically and dynamically stable in two geometrical arrangements, namely, buckled and puckered configurations. The puckered monolayer is a metal, whereas the buckled monolayer is a semimetal. Interestingly, the electronic band structure of the buckled configuration possesses a linear dispersion and a Dirac cone at the Fermi level around the high-symmety K point in the reciprocal lattice. Thus, at low-energy excitation (up to 105 meV), the charge carriers in this system behave as massless Dirac fermions. Detailed analysis of partial density of state indicates that the 2pz orbital of C atoms contributes significantly to the states which form the linear dispersion and hence the Dirac cone around the Fermi level. This suggests the existence of a strong correlation between the linear dispersion (Dirac cone) and sp2-like hybridization of the orbitals of C atom. Thus the electronic properties of CAs3 monolayer are similar to those of graphene and other group-IV based monolayers like, silicene and germanene. In addition, we have also investigated the influence of mechanical strain on the properties of CAs3 monolayer. Our results indicate that the monolayer possesses linear dispersion in the electronic band structure for a wide range of mechanical strain from −12% to 20%, though the position of Dirac point may not lie exactly at the Fermi level. Finally, we wish to point out that CAs3 monolayer belongs to the class of Dirac materials where the behavior of particles, at low-energy excitations, is characterized by the Dirac-like Hamiltonian rather than the Schrodinger Hamiltonian.
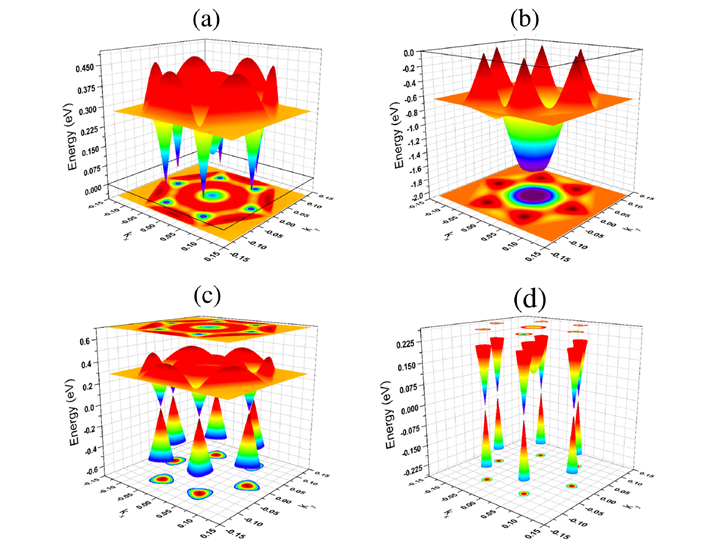 Figure 8 Three-dimensional plots of electronic band structure of CAs3 monolayer in buckled configuration: (a) lowest unoccupied conduction and (b) highest occupied valence bands which are close to the Fermi level. Combined plots of these two bands are given (c) and (d) in two different energy ranges. Presence of Dirac cones at the Fermi level is clearly visible in (d).
Figure 8 Three-dimensional plots of electronic band structure of CAs3 monolayer in buckled configuration: (a) lowest unoccupied conduction and (b) highest occupied valence bands which are close to the Fermi level. Combined plots of these two bands are given (c) and (d) in two different energy ranges. Presence of Dirac cones at the Fermi level is clearly visible in (d).
(7) Isoelectronically substituted group-III based monolayers
Using density functional theory based electronic structure calculations, we carry out detailed investigation on geometric and electronic properties of group-III atom (B, Al, and Ga) based monolayers, namely borophene, aluminene, and gallinene, in β12 configuration which is one of the stable structures for borophene. The results of our calculations indicate that all these monolayers form energetically stable systems and these are all metallic in nature since they possess finite density of states at the Fermi level. Analysis of optimized geometric structures reveals that while borophene in β12 structure stabilizes in planar structure, the geometries of the remaining monolayers are nonplanar. In the case of borophene, there is a signature of crossing of two linear bands (which form Dirac cones) at about 2 eV and 0.5 eV above the Fermi level. Similar features have also been observed in electronic band structures of both aluminene and gallinene. Interestingly, one of the Dirac cones in these monolayers lies much closer to the Fermi level compared to the borophene case. The Fermi velocities around these Dirac cones are also higher and comparable to that in graphene. We further study the effect of isoelectronic substitution at one of the inequivalent sites (C site) of the β12 configuration for these three monolayers. Our results show that borophene still retains its planar geometry following the substitution. Interestingly, in all these cases, crossing of linearly dispersing bands (Dirac cones) is observed nearer to the Fermi level compared to the pristine systems. Our study suggests that isoelectronic substitutions may be useful in tuning the electronic properties, in particular linear dispersion, of monolayer systems.
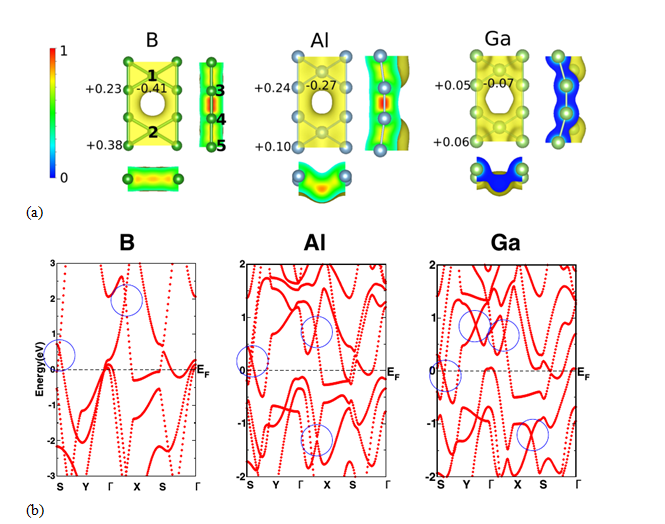 Figure 9 (a) Spatial distribution of valence charge density for the group-III based monolayers. 0 and 1 in the bar scale represents the charge deficit and the charge rich region, respectively. Bader charges on symmetrically inequivalent atoms [1(2), 3(4), and 5, respectively, at the sites A, B, and C) are shown.(b) The electronic band structures of the group-III based monolayers in β12 structure. The blue circles indicate the locations of linear band crossings (Dirac-like cones). The Fermi energy is set to 0 eV.
Figure 9 (a) Spatial distribution of valence charge density for the group-III based monolayers. 0 and 1 in the bar scale represents the charge deficit and the charge rich region, respectively. Bader charges on symmetrically inequivalent atoms [1(2), 3(4), and 5, respectively, at the sites A, B, and C) are shown.(b) The electronic band structures of the group-III based monolayers in β12 structure. The blue circles indicate the locations of linear band crossings (Dirac-like cones). The Fermi energy is set to 0 eV.
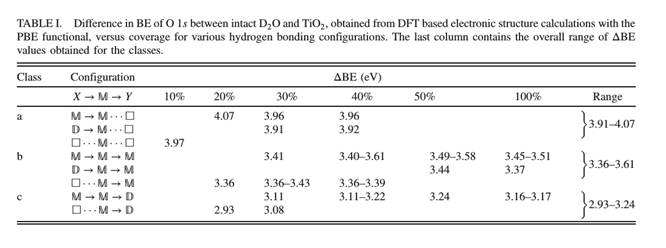
(8) Core-Level Binding Energy Reveals Hydrogen Bonding Configurations of Water Adsorbed on TiO2(110) Surface
Using x-ray photoelectron spectroscopy of the oxygen 1s core level, the ratio between intact (D2O) and dissociated (OD) water in the hydrated stoichiometric TiO2(110) surface is determined at varying coverage and temperature. In the submonolayer regime, both the D2O∶OD ratio and the core-level binding energy of D2O (ΔBE) decrease with temperature. The observed variations in ΔBE are shown with density functional theory to be governed crucially and solely by the local hydrogen bonding environment, revealing a generally applicable classification and details about adsorption motifs.
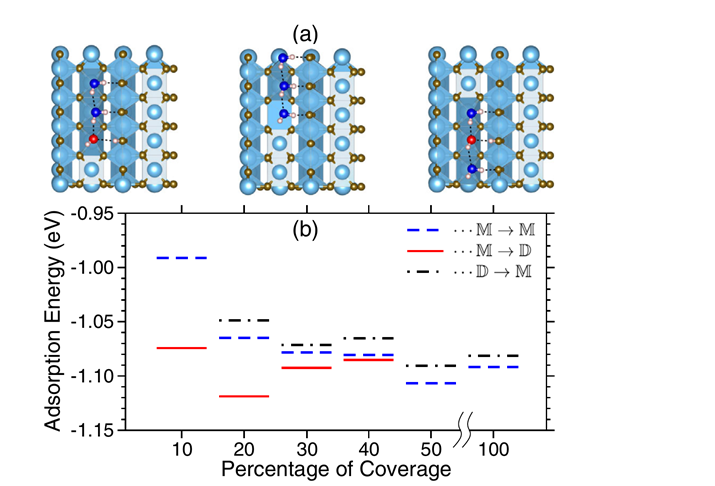 Figure 10 (a) Top view of three possible geometric configurations (M → D, M → M, and D → M) for 30% coverage of water molecules on TiO2(110) (b) Adsorption energy versus coverage.
Figure 10 (a) Top view of three possible geometric configurations (M → D, M → M, and D → M) for 30% coverage of water molecules on TiO2(110) (b) Adsorption energy versus coverage.
(9) Strong Field Quantum electrodynamics: Prediction of a Novel Dynamical StageField induced Phase transition of vacuum state
The conjecture of “false vacuum” is a holy grail of quantum electrodynamics. A possible testing ground for this may come up at XFEL, DESY, Hamburg, Germany through the production of particle-antiparticle pairs from the vacuum fluctuation in a sufficiently strong time-dependent electric field (comparable to Schwinger’s critical field ES = 1.32 × 1018 V/m). Such a pair creation can be viewed as a field induced phase transition (FIPT) via the t-non invariant vacuum state because of the non-stationary Hamiltonian wherein the spontaneous symmetry breaking of the vacuum state takes place under time inversion and consequently electron-positron pairs are generated as the massive analogue of Goldstone bosons. The dynamics of FIPT of the vacuum state interacting with time dependent Sauter pulse is studied to analyse different evolution stages of the order parameter e.g., quasi electron positron plasma (QEPP), transient, and residual electron-positron plasma (REPP) stages. We attribute the appearance of the transient stage to the nonlinear coupling in the differential equations governing the dynamics of the phase and the modulus of the order parameter. The onset of the rapid oscillations in the modulus is shown to be associated with the abrupt change in the phase of the order parameter in the transient stage. FIPT is also studied for multi-sheeted Sauter pulse with linear and quadratic frequency chirp. The formation of a novel pre-transient region due to the quadratic frequency chirping is observed in the accelerating part of the QEPP stage before the electric field attains the maximum value. Increase in the quadratic chirp is shown to strongly enhance the pair production rate by five orders of magnitude.
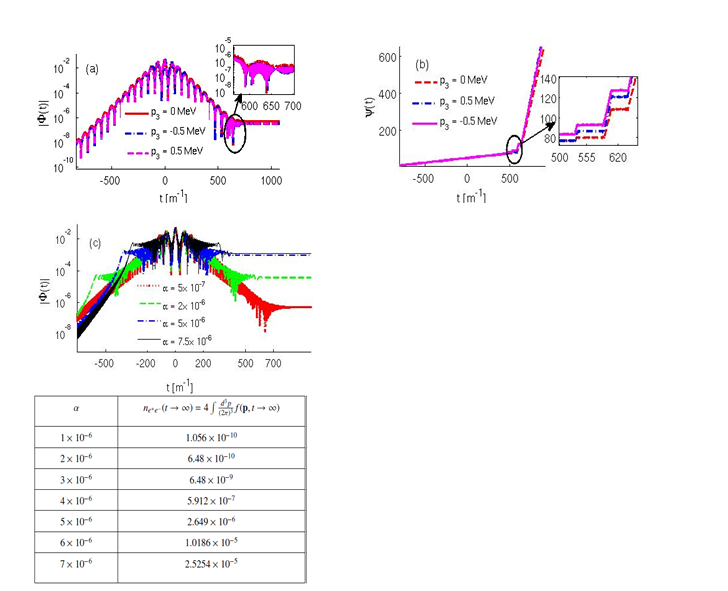 Figure 11 Dynamical characterization of field induced phase transition of vacuum state. (a) Evolution of modulus of complex order parameter |Φ(t)| (b) Evolution of phase of the order parameter ψ(t) for a Sauter pulse with 5 subcycle oscillations for different values of longitudinal momentum p3. Note that the onset of transients in |Φ(t)| is accompanied by the abrupt change in the phase ψ(t). (c) Evolution of |Φ(t)| for the Sauter pulse with quadratic frequency chirp of strength α for longitudinal momentum p¬3 = 0. A pre-transient stage is clearly seen in the rising part of the pulse. The table on right bottom panel shows the enhancement in the total number of electron-positron pairs with the increase in the strength of quadratic frequency chirping. The peak electric field E0 = 0.1ES, and the pulse duration τ = 100 unit. Time is shown in the electron mass unit.
Figure 11 Dynamical characterization of field induced phase transition of vacuum state. (a) Evolution of modulus of complex order parameter |Φ(t)| (b) Evolution of phase of the order parameter ψ(t) for a Sauter pulse with 5 subcycle oscillations for different values of longitudinal momentum p3. Note that the onset of transients in |Φ(t)| is accompanied by the abrupt change in the phase ψ(t). (c) Evolution of |Φ(t)| for the Sauter pulse with quadratic frequency chirp of strength α for longitudinal momentum p¬3 = 0. A pre-transient stage is clearly seen in the rising part of the pulse. The table on right bottom panel shows the enhancement in the total number of electron-positron pairs with the increase in the strength of quadratic frequency chirping. The peak electric field E0 = 0.1ES, and the pulse duration τ = 100 unit. Time is shown in the electron mass unit.
(10) Charge Transport in semicrystalline polymer films: Negative field dependence of mobility can occur even without positional disorder
A new model for simulating the charge transport properties of semicrystalline polymer using Monte Carlo simulation is proposed. It is used to explain the experimentally observed field and temperature dependence of mobility in Poly(3-hexylthiophene-2,5-diyl) (P3HT) thin films. This study also provides a new physical insight to the origin of much debated negative field dependence of mobility (NFDM) observed at low electric field strengths in P3HT thin films which earlier models could not explain. Contrary to the existing belief, it is clearly established that NFDM can be observed even in the absence of any positional disorder.
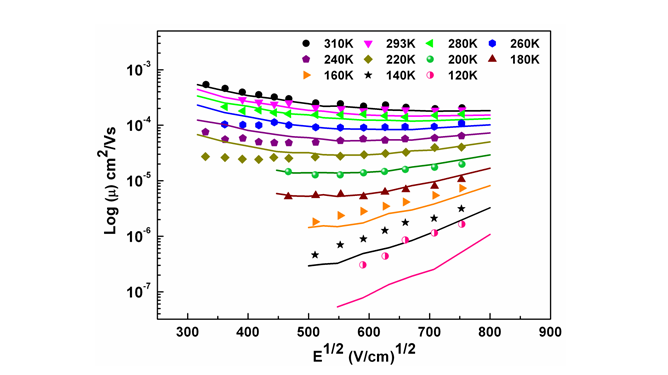 Figure 12. Modeling the experimental field dependence of mobility data at different temperature reported by Mozer and Sariciftci, Chem. Phys. Lett. 389, 438 (2004). Lines represents the fit to the experimental data shown in symbols.
Figure 12. Modeling the experimental field dependence of mobility data at different temperature reported by Mozer and Sariciftci, Chem. Phys. Lett. 389, 438 (2004). Lines represents the fit to the experimental data shown in symbols.
(11) Molecular-scale understanding of the mechanism of solution-mediated nucleation and growth of crystalline materials
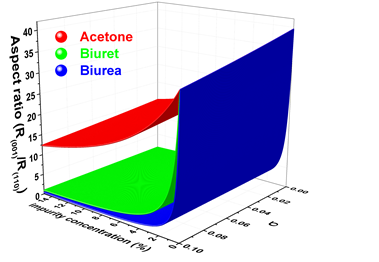
Molecular-scale understanding of the mechanism of solution-mediated nucleation and growth of crystalline materials in the presence of growth inhibitor together with the process parameters continues to attract interest of the scientific community though much headway has been made in recent years. The role of different growth additives like acetone, biuret and biurea has been investigated to study aqueous crystallization of the urea crystal. The various low energy configurations of additive-surface have been obtained using evolutionary based algorithm. The structure and adsorption energies of different configurations of additives at different faces of urea crystal have been calculated using periodic dispersion corrected density functional method. The calculated adsorption energies of different auxiliaries are employed to examine the role played by these auxiliaries during aqueous crystallization of urea crystal containing small amounts of additives. Our results show that the growth of (110), (111) and faces are nearly unaltered by the addition of moderate amounts of acetone as it has lower adsorption energies with the surfaces of these faces. Nevertheless, the presence of acetone in the solution reasonably impedes the growth of (001) face. Addition of biuret or biurea in the solution lead to higher adsorption energy at (001) and (111) faces. Consequently the low concentration of these additives severely obstructs the growths of (001) and (111) faces as most of the adsorption sites are occupied by these additives. On the other hand, these additives are weakly adsorbed at (110) face and, hence, the growth of (110) face largely remains unaltered. Moreover, unlike to biuret, biurea considerably inhibit the growth of face. Our results are in agreement with the experimental and computational results reported in the literature.
|